In short...
It
is generally accepted that many functional proteins do not have
well-defined folded structures. These so-called intrinsically
disordered proteins (IDPs) are encoded abundantly in the human genome
and are involved in a variety of biological process including cell
signalling, cell cycle control, molecular recognition, nucleic acid
transcription and replication, as well as the development of
neurodegenerative diseases and cancer. The studies of IDPs is an
emerging field of research and general rules for describing their
conformational behaviour and mechanisms are still missing. Thus,
expanding the amount of experimental data from different systems as
well as developing new techniques to characterize their properties are
essential to improve our knowledge about this family of proteins.
We
are interested in using the state-of-the-art nuclear magnetic resonance
techniques and other biophysical methods in combination with novel
computational modelling to explore the structural propensity and
dynamics of IDPs and the mechanism of their interaction with other
proteins or nucleic acids at atomic resolution.
Intrinsically disordered proteins
After
a decade in the post-genome era, the determination of the functions of
proteins encoded in DNA sequences is still one of the major challenges.
It is a widely accepted concept that the function of a protein is
determined by its three-dimensional structure. Numerous protein
structures with their functional interpretations deposited in the
Protein Data Bank (PDB) over last fifty years strongly support this
idea. However, this structure-function paradigm has been reassessed
extensively in recent years. From bioinformatics studies, intrinsically
disordered proteins have been shown to be amply present in all kingdoms
of life. It is estimated that approximately 50% of mammalian proteins
contain long disordered regions (more than 30 residues), and
approximately 25% of their proteins are expected to be fully disordered
under physiological conditions. The lack of folded structure provides
several advantages such as having a larger solvent exposed surface to
enhance the chance of interacting with binding partners via the
so-called “fly-casting” mechanism, as well as allowing them to act as
scaffolds by interacting with different proteins. One of the most
intriguing aspects of disordered proteins is that they often undergo
structural transitions from disordered to folded forms upon binding to
their physiological partners. This folding-upon-binding mechanism opens
a new view of protein-protein and protein-DNA/RNA interactions. In
spite of the advantages of being unstructured, the disorderedness of
some of these proteins also leads to disease related aggregation or
fibrillization. It is also estimated from bioinformatics studies that
about 80% of cancer-associated proteins contain consecutive disordered
regions. This new class of proteins is now mostly termed intrinsically
disordered proteins (IDPs) or intrinsically disordered regions (IDRs)
of structured proteins. With those key studies elucidating the
importance of IDPs, “protein disorder” has become an emerging research
field. From the accumulating amount of studies, it is now generally
believed that IDPs play key roles in many physiological processes,
including cell signalling, cell cycle control, molecular recognition,
nucleic acid transcription and replication, as well as in the
development of neurodegenerative diseases, cardiovascular diseases,
amyloidoses, and type II diabetes.
Physiological and
biochemical results have drawn our attention to the importance of IDPs,
but several aspects about the mechanisms of IDP function are still
unknown: How are IDPs recognized by the partner proteins in the absence
of a folded structure? Does any specific pre-recognition conformation
exist with their flexible nature? Can we derive a general rule to
understand the conformational behaviour of these proteins from the
primary sequence? In other words, can we predict the functions and
mechanisms of IDPs from primary sequence? Insights into the dynamics
and conformational propensities of these proteins at the atomic level
will be a critical step on the way to answer these questions.
Conventional approaches for structure determination or characterization
is less feasible due to the structural heterogeneity of IDPs. Novel
biophysical methods and computational models, therefore, become
essential to overcome their rapidly inter-converting nature.
Our
group is interested in using, nuclear magnetic resonance (NMR)
spectroscopy, giving specific information for almost all atoms with
minimal interference, to characterize IDPs. Particularly, two of the
latest developed NMR techniques, residual dipolar couplings (RDCs) and
paramagnetic relaxation enhancements (PREs), which are extremely
sensitive to local conformational sampling and transient long-range
interaction in unstructured proteins, will be applied to those systems
studied. Other biophysical methods such as small angle X-ray scattering
(SAXS), circular dichroism spectroscopy, and fluorescence spectroscopy
will also be used as complementary methods. In addition, due to the
heterogeneity of IDPs, a statistically significant computational model
will be used to characterize the structural propensities of the IDPs.
We are using experimental data as constraints to obtain representative
conformational ensembles of IDPs. We are also developing new methods
hopefully to predict the function of IDPs solely on the basis of
primary sequence. We hope studies carried on in our group will improve
our understanding of the structural dynamics, conformational behaviour,
related biological processes, and the onset of pathological aggregation
or fibrillization of IDPs.
Nuclear Magnetic Resonance Spectroscopy
NMR
spectroscopy, giving specific information for almost all atoms with
minimal interference, is one of the most powerful tools for
experimental characterization of disordered proteins. In addition to
those regularly measured parameters (chemical shifts, scalar couplings,
nuclear Overhauser effects, and relaxation rates), two more recently
developed experimental parameters, residual dipolar couplings (RDCs)
and paramagnetic relaxation enhancements (PREs), will also be applied
to probe the local conformational sampling and long-range distance
information in IDPs.
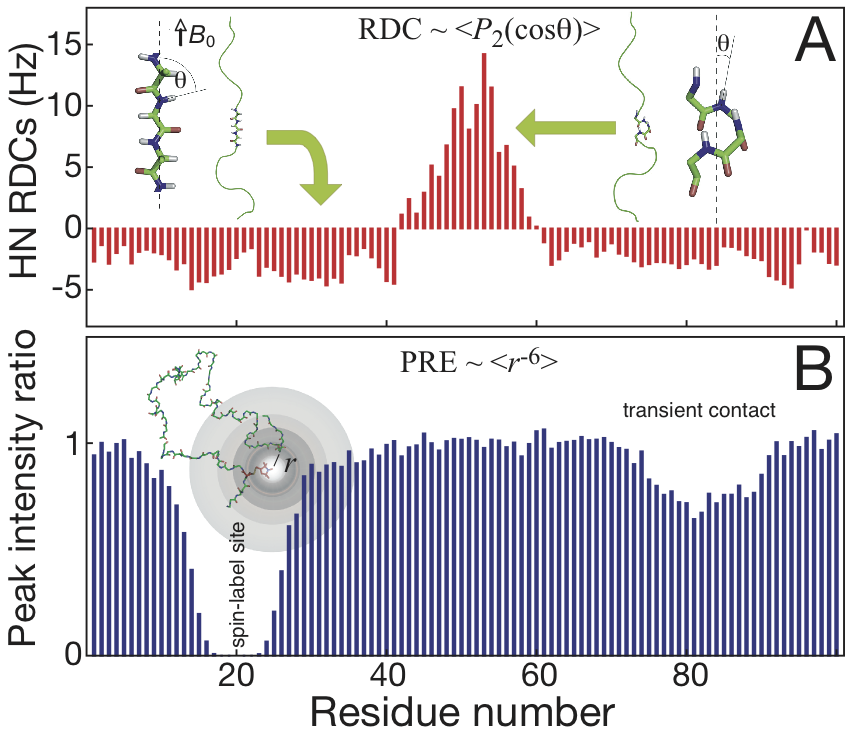
Residual dipolar couplings
The
size of RDCs can be calculated very precisely as ensemble and time
averages from the well-understood geometry dependence of
nucleus-nucleus dipolar interactions. In solution, this interaction
vanishes due to molecular tumbling. However, a small part of the
dipolar interaction (denoted as residual dipolar coupling) can be
re-introduced by dissolving the protein molecules in weak alignment
media such as stretched polyacrylamide gel or bicelles. As an
illustrative example, the RDCs between amide nitrogen and proton (NH)
are negative on the elongated part of a disordered protein because the
angle between the NH vector and the external magnetic field is close to
perpendicular, leading to the cosine function of such angle in the
second-order Legendre polynomial to an extreme (the molecule supposed
to be aligned parallel to the magnetic field). In contract, if there is
a significant helical component populated, the angle would be close to
zero leading to positive RDC values. Therefore, RDCs are extremely
useful for local conformational studies even in the case of transiently
populated structural propensities.
Paramagnetic relaxation enhancement
In
contrast to RDCs which reports on local conformational sampling, PREs
provide information about transient long-range contacts for inter- or
intra- protein interactions. PREs can be observed after introducing a
suitable paramagnetic tag, such as commercially available nitroxide
MTSL or lanthanide chelating tags. Because the gyromagnetic ratio of
the electron spin is over 600 times larger than the proton spin, the
observed line broadening due to paramagnetic relaxation enhancement
provides long-range probes of distances over 25 Å even if the contacts
are weakly or transiently populated. In addition to using NMR signal
line-broadening to estimate PREs as commonly used, explicit relaxation
rates for different types of nuclei will also be recorded explicitly to
reduce the uncertainties from the complexity of correlation times in
unfolded proteins, and to provide sufficient and precise distance
information for the characterization of the IDPs.
Computational modelling
Statistical coil model and constrained subensemble selection

The
so-called statistical coil model consists of an ensemble of structures
in which the backbone dihedral angles sample amino acid-specific energy
potentials based on their occurrence in the non-α-helical and
non-β-sheet regions of highly resolved X-ray structures. An extremely
efficient algorithm, flexible-Meccano,
can be used to construct such model. This approach has been
demonstrated to provide theoretical RDCs that compare well with
experimental values in several cases. The deviation between predicted
and experimental values is indicative of the presence of long-range
contacts or residual. Furthermore, using a genetic algorithm, Asteroids, developed in Blackledge's group, a subensemble of structures that fulfils experimental data can be selected from flexible-Meccano
generated pool. Residue-specific information of IDPs can be revealed
from the selected subensembles using experimental observables such as
RDCs, PREs, CSs and SAXS.
Restrained molecular dynamics simulation
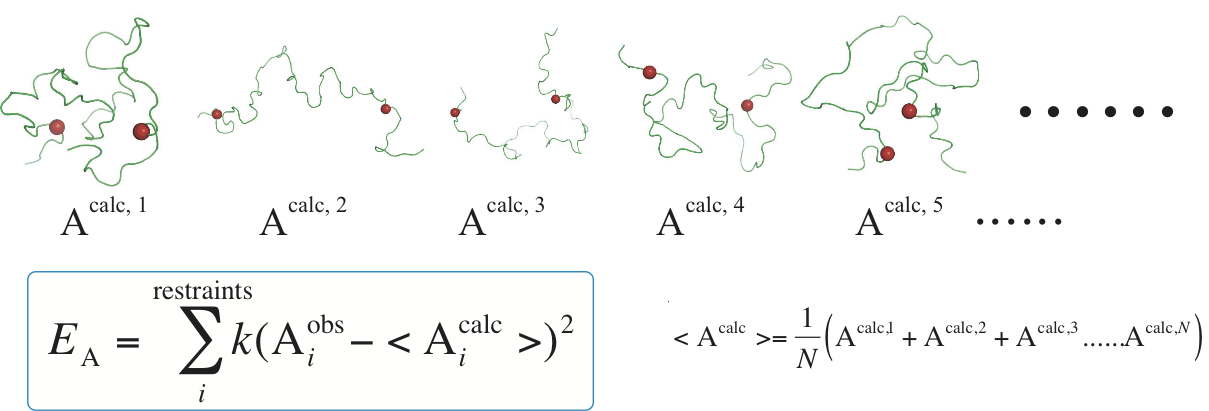
Alternative
to using conformational sampling and selecting method, MD simulation
gives a route to dynamic properties and energy evolution. Due to the
lack of computational power and underdevelopment of force field for
unstructured proteins, restraint-free MD simulation is still
challenging. Currently, simulation with assistance of experimental
observables is a more feasible approach. In restrained MD simulation, a
pseudo-energy potential term is added to the total energy function of
the simulated system to minimize the difference between calculated
values and experimental data. In addition, due to the heterogeneity of
unstructured systems, a single conformer is not sufficient and not
realistic to fulfill all experimental restraints. Therefore, a replica
of structures is running in parallel and only the calculated values
averaged over all conformers are necessary to target to experimental
restraints.
Other biophysical techniques
Small angle X-ray scattering
SAXS
has been used to characterize the shape of interacting proteins and
overall dimensions of unfolded peptide chains. Unlike crystallography,
sample prepared for SAXS methods is in solution similar to experiments
conducted in NMR spectroscopy. Accordingly, SAXS is widely applied as a
complementary method with NMR studies. The National Synchrotron
Radiation Research Center has a beamline (BL23A) specifically dedicated
for SAXS studies, providing a convenient access for SAXS measurement.
Spectroscopic techniques
Fluorescence
and circular dichroism (CD) spectroscopy provide immediate assay of
protein disorder. Far UV-CD is also sensitive to the poly-proline II
helix conformation often populated in IDPs. These techniques will be
used a preliminary check of the level of protein disorder.
References
General news/books about IDPs
- T. Chouard. Breaking the protein rules. Nature, 471: 151-3, 2010.
- J. Schnabel. The dark side of proteins. Nature, 464: 828-9, 2010.
Scientific reviews/books about IDPs
- P. Tompa. Structure and function of intrinscially disordered proteins. CRC Press, 2010.
- V. N. Uversky and A. K. Dunker. Understanding protein non-folding. Biochim Biophys Acta, 1804(6):1231–64, 2010.
- A. K. Dunker, I. Silman, V. N. Uversky, and J. L. Sussman. Function and structure of inherently disordered proteins. Curr Opin Struct Biol, 18:756–64, 2008.
- P. E. Wright and H. J. Dyson. Linking folding and binding. Curr Opin Struct Biol, 19:31–8, 2009.
- C. M. Dobson. Protein folding and misfolding. Nature, 426:884–90, 2003.
- R.
Schneider, J.-R. Huang, M. Yao, G. Communie, V. Ozenne, L. Mollica, L.
Salmon, M. R. Jensen, and M. Blackledge. Towards a robust description
of intrinsic protein disorder using nuclear magnetic resonance
spectroscopy. Mol Biosys, 8(1):58–68, 2012.
Scientific articles/reviews about NMR
- M. Blackledge. Recent progress in the study of biomolecular structure and dynamics in solution from residual dipolar couplings. Prog Nucl Magn Reson Spectrosc, 46(1):23–61, 2005.
- J. H. Prestegard, C. M. Bougault, and A. I. Kishore. Residual dipolar couplings in structure determination of biomolecules. Chem Rev, 104(8):3519–40, 2004.
- G.
M. Clore and J. Iwahara. Theory, practice, and applications of
paramagnetic relaxation enhancement for the characterization of
transient low-population states of biological macromolecules and their
complexes. Chem Rev, 109(9):4108–39, 2009.
- G. Otting. Protein NMR using paramagnetic ions. Annu Rev Biophys, 39:387–405, 2010.
